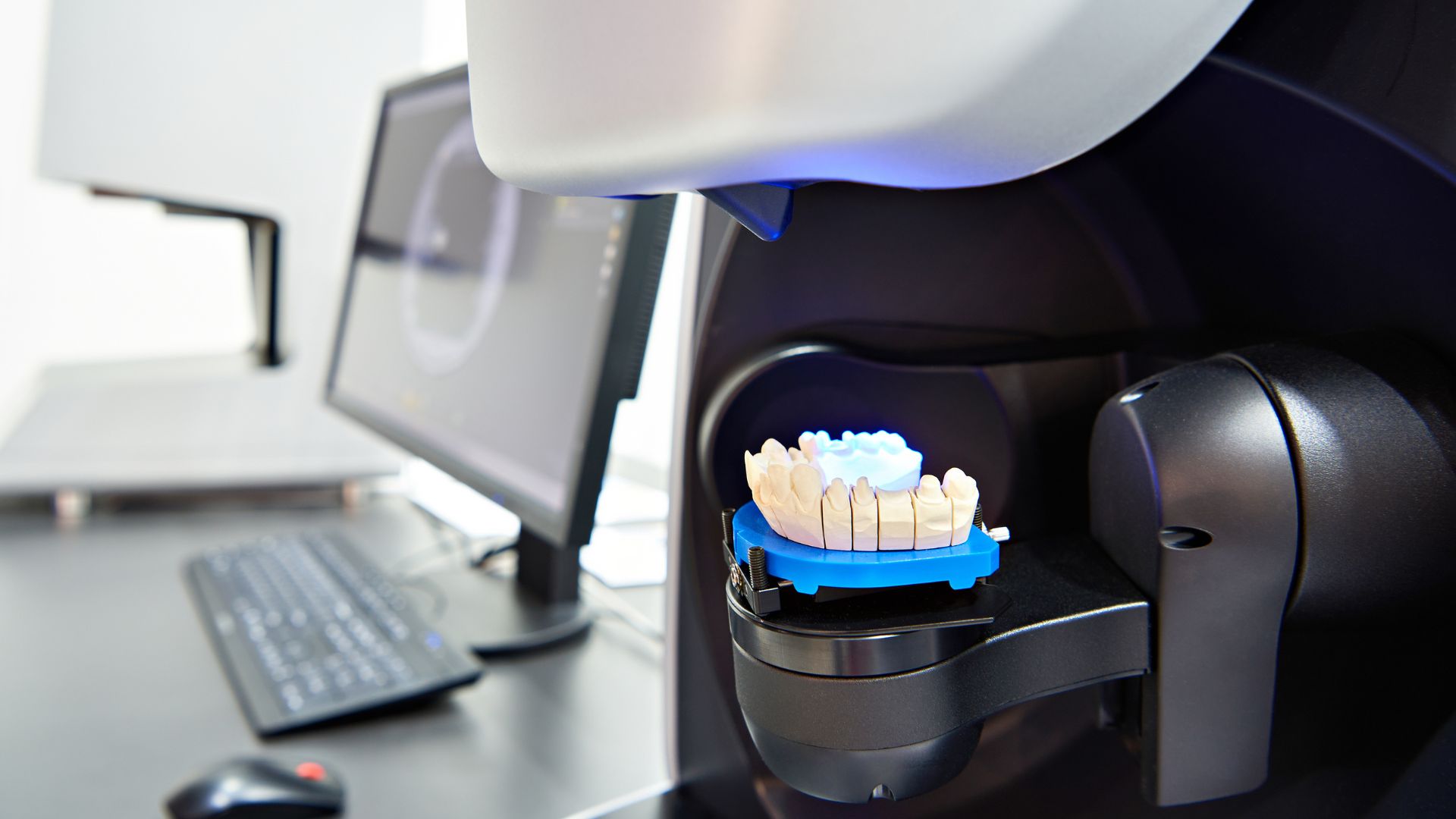
Dental impression materials & digital scanners are widely used in restorative & orthodontic treatments, ensuring precision in dental prosthetics & aligners. Because these devices play a crucial role in patient outcomes, the FDA enforces strict regulatory controls to ensure safety & accuracy. Manufacturers marketing impression materials or intraoral scanners in the US must comply with medical device regulations, classification requirements & labeling standards. Non-compliance can lead to Import Alerts, detentions & enforcement actions.
How the FDA Classifies Dental Impression Products
FDA classification is based on risk level & device functionality:
- Class I (Low Risk): Basic impression trays & mixing spatulas. Typically exempt from 510(k) but require Establishment Registration & Medical Device Listing.
- Class II (Moderate Risk): Alginate, PVS & other elastomeric impression materials. These require 510(k) clearance.
- Class II (Moderate Risk): Digital intraoral scanners & CAD/CAM impression systems. Also require 510(k) clearance.
Steps to Enter the US Market
Manufacturers must complete several regulatory steps before distributing impression materials or scanners:
- Establishment Registration: Required annually for all manufacturers & importers.
- Medical Device Listing: Each product must be listed under the registered establishment.
- UDI Compliance: Most digital scanners require a Unique Device Identifier for traceability.
- Labeling & Advertising Compliance: Packaging & claims must meet FDA regulations.
Common Compliance Challenges & Solutions
Even experienced manufacturers can face unexpected regulatory hurdles. The following examples highlight the importance of preparation:
Case Study: Digital Impression Scanner Detained Due to UDI Non-Compliance
A manufacturer attempted to launch an intraoral scanner in the US but was detained due to missing UDI labeling:
- Shipments delayed while compliance updates were implemented.
- Packaging redesigned & UDI data submitted to GUDID.
- Regulatory consultants engaged to prevent future detentions.
Case Study: Incorrect Classification of a New Impression Material
A company misclassified a self-curing impression material as Class I. The FDA reclassified it as Class II:
- Product launch delayed while conducting safety testing.
- Additional costs incurred for regulatory submissions.
- Filed a 513(g) request to clarify classification before future development.
Regulatory Considerations for Manufacturers
- FDA User Fees: Required annually; Small Business Fee Assistance may apply.
- Import Alerts: Violations can result in blocked shipments.
- Certificate to Foreign Government (CFG): Needed for international exports.
- Health Canada Licensing: MDEL required to distribute in Canada.
Maintaining Compliance After Market Entry
Approval is only the beginning. Ongoing responsibilities include:
- eMDR Submissions: Report device-related malfunctions or adverse events.
- FOIA Requests: Access regulatory data on comparable devices.
- Medical Device Master File: Streamlines future approvals.
- Ongoing Regulatory Consulting: Ensures alignment with FDA updates.
Setting the Stage for Regulatory Success
Bringing dental impression materials & scanners to the US market requires more than approval—it demands continuous regulatory alignment. Manufacturers that correctly classify devices, follow UDI & labeling rules & engage regulatory support will enter the market with confidence & maintain long-term success.
Medical Devices
Latest Articles




Author
