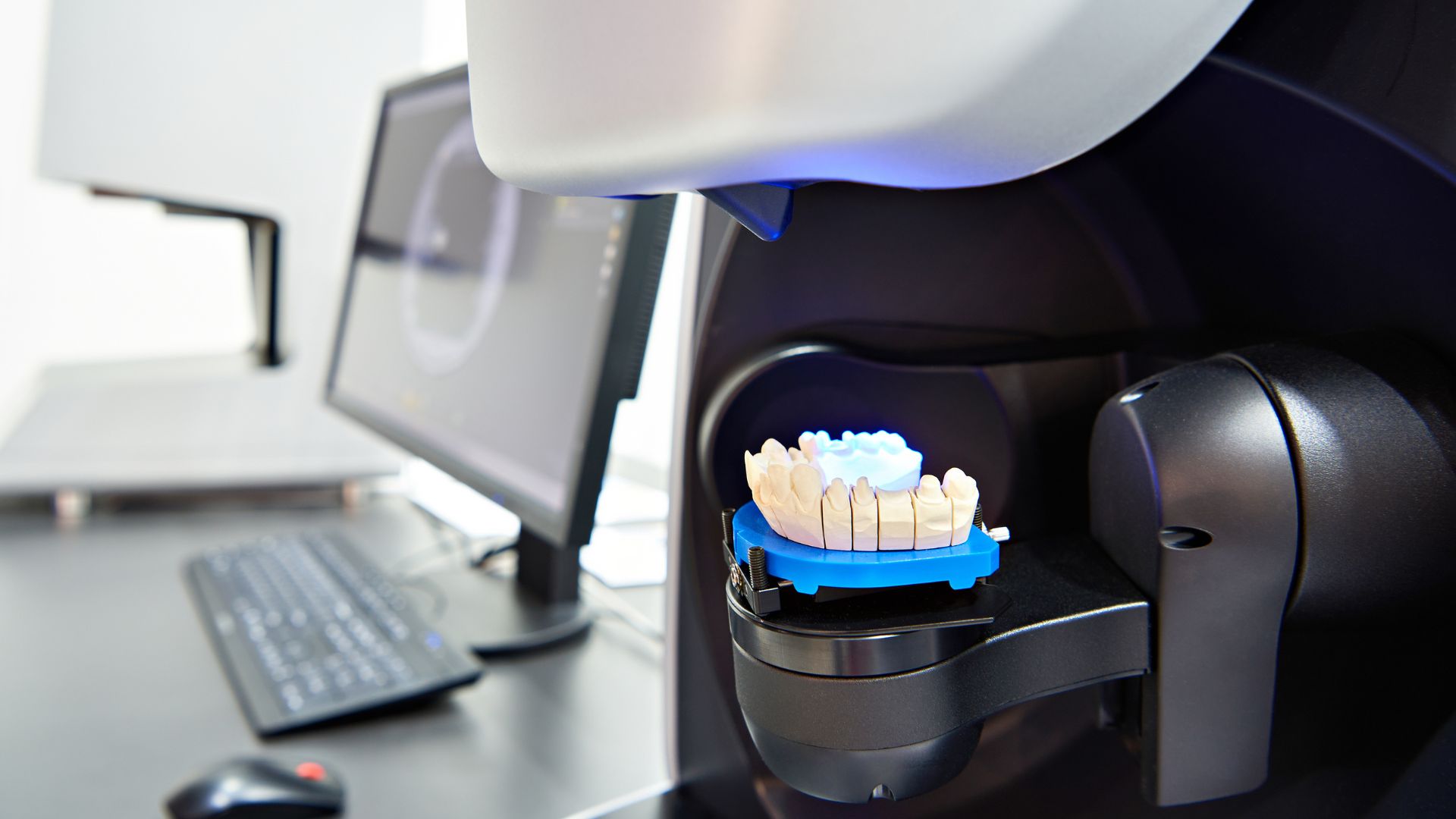
Os materiais de moldagem dentária e scanners digitais são amplamente usados em tratamentos restauradores e ortodônticos, garantindo precisão em próteses dentárias e alinhadores. Como esses dispositivos desempenham um papel crucial nos resultados dos pacientes, a FDA impõe controles regulatórios rigorosos para garantir a segurança e a precisão. Os fabricantes que comercializam materiais de impressão ou scanners intraorais nos EUA devem cumprir os regulamentos de dispositivos médicos, requisitos de classificação e padrões de rotulagem. A não conformidade pode levar a alertas de importação, detenções e ações de execução.
Como a FDA classifica os produtos de impressão dental
A classificação da FDA é baseada no nível de risco e na funcionalidade do dispositivo:
- Classe I (baixo risco): Bandejas de impressão básicas e espátulas de mistura. Normalmente isento de 510(k), mas exige registro de estabelecimento e listagem de dispositivos médicos.
- Classe II (risco moderado): Alginato, PVS e outros materiais de moldagem elastoméricos. Isso requer uma folga de 510(k).
- Classe II (risco moderado): Scanners intraorais digitais e sistemas de impressão CAD/CAM. Também requer espaço livre 510(k).
Etapas para entrar no mercado dos EUA
Os fabricantes devem concluir várias etapas regulatórias antes de distribuir materiais de impressão ou scanners:
- Registro de estabelecimento: Exigido anualmente para todos os fabricantes e importadores.
- Listagem de dispositivos médicos: Cada produto deve ser listado no estabelecimento registrado.
- Conformidade com UDI: A maioria dos scanners digitais exige um identificador de dispositivo exclusivo para rastreabilidade.
- Conformidade de rotulagem e publicidade: Embalagens e reivindicações devem atender aos regulamentos da FDA.
Desafios e soluções comuns de conformidade
Até mesmo fabricantes experientes podem enfrentar obstáculos regulatórios inesperados. Os exemplos a seguir destacam a importância da preparação:
Estudo de caso: Scanner de impressão digital retido devido a não conformidade com UDI
Um fabricante tentou lançar um scanner intraoral nos EUA, mas foi detido devido à falta de rotulagem de UDI:
- Remessas atrasadas enquanto atualizações de conformidade eram implementadas.
- Dados de UDI e reprojetados da embalagem enviados ao GUDID.
- Consultores regulatórios contratados para evitar detenções futuras.
Estudo de caso: Classificação incorreta de um novo material de impressão
Uma empresa classificou erroneamente um material de impressão autorrecuperável como Classe I. A FDA o reclassificou como Classe II:
- Lançamento do produto atrasado durante a realização de testes de segurança.
- Custos adicionais incorridos para submissões regulatórias.
- Arquivada uma solicitação 513(g) para esclarecer a classificação antes do desenvolvimento futuro.
Considerações regulatórias para fabricantes
- Taxas de usuário da FDA: Exigido anualmente; pode ser aplicada a assistência para pequenas empresas.
- Alertas de importação: Violações podem resultar em remessas bloqueadas.
- Certificado para governo estrangeiro (CFG): Necessário para exportações internacionais.
- Licenciamento da Health Canada: O MDEL deve ser distribuído no Canadá.
Manutenção da conformidade após entrada no mercado
A aprovação é apenas o começo. As responsabilidades contínuas incluem:
- Envios de eMDR: Relatar mau funcionamento ou eventos adversos relacionados ao dispositivo.
- Solicitações FOIA: Acesse dados regulatórios em dispositivos comparáveis.
- Arquivo mestre de dispositivos médicos: Simplifica as aprovações futuras.
- Consultoria regulatória contínua: Garante o alinhamento com as atualizações da FDA.
Preparando o cenário para o sucesso regulatório
Levar materiais e scanners de moldagem dentária para o mercado dos EUA exige mais do que aprovação, exige alinhamento regulatório contínuo. Os fabricantes que classificam corretamente os dispositivos, seguem as regras de UDI e rotulagem e envolvem suporte regulatório entrarão no mercado com confiança e manterão o sucesso a longo prazo.
Autor
